Case Study
TGX – Toxicogenomics-based prediction and mechanism identification
Summary
In this case study a transcriptomics-based hazard prediction model for identification of specific molecular initiating events (MIE) was foreseen based on (A) top-down and (B) bottom-up approaches.
The MIEs can include, but are not limited to: (1) Genotoxicity (p53 activation), (2) Oxidative stress (Nrf2 activation), (3) Endoplasmic Reticulum Stress (unfolded protein response), (4) Dioxin-like activity (AhR receptor activation), (5) HIF1 alpha activation and (6) Nuclear receptor activation (e.g. for endocrine disruption).
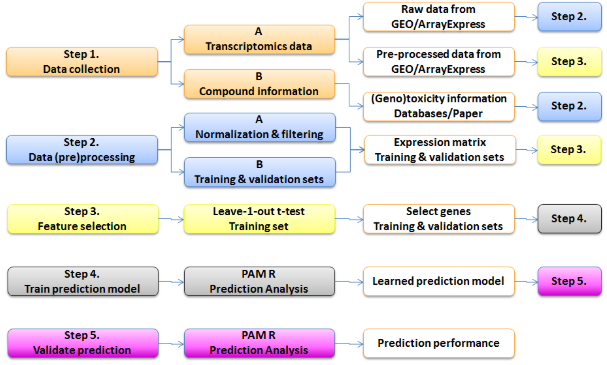
This case study focussed on two top-down approaches for genotoxicity prediction. The first approach resulted in the creation of a Nextflow-based workflow from the publication “A transcriptomics-based in vitro assay for predicting chemical genotoxicity in vivo” by Magkoufopoulou et al. (2012), thereby reproducing their work as proof of principle. The workflow for one of the prediction models described in the publication is shown in the Figure below.
The Nextflow-based workflow has been translated into a more generic approach, especially for step 1, forming the basis of the second top-down approach. In this approach transcriptomics data together with toxicological compound information were collected from multiple toxicogenomics studies and used for building a metadata genotoxicity prediction model.
Objectives
- Creation of prediction models based on differentially regulated genes (top-down approach);
- Using knowledge of stress response pathways to integrate data sets for their activation or inhibition (bottom-up approach).
These two use cases are relevant for the top-down approaches:
- Reproducing the prediction models published by Magkoufopoulou et al. (2012);
- Advanced predictions using as much data as possible from the diXa data warehouse and other repositories giving free access to the data.
Risk assessment framework
This case study is associated with all 3 tiers of the selected framework and in particular the following steps:
- Collection of support data;
- Identification of analogues / suitability assessment and existing data;
- Mode of Action hypothesis generation.
Databases and tools
Databases:
- diXa data warehouse (carcinoGENOMICs, Predict-IV), TG-GATEs, ArrayExpress/GEO, BioStudies.
Tools:
- Top-down: data normalisation tools, prediction tools such as Caret;
- Bottom-up: ToxPi.
Technical implementation
Integration with other case studies is needed. TGX acquires information and data from the DataCure case study as well as through the services ToxPlanet and ToxicoDB of the Implementation Challenge winners Toxplanet and UHH, respectively. The results of TGX can feed into SysGroup, AOPLink and ModelRX.
Currently available services:
- Service to run Nextflow pipelines
- Service type: Service, Workflow, Software
Transcriptomics data from human, mouse, rat in vitro liver models:
- Repository for transcriptomics data from multiple in vitro human, rat and mouse toxicogenomics projects
- Service type: Database / data source
Outcomes
Outcome from this case study provides workflows for obtaining data which are suited for developing toxicity prediction models. This resulted in two top-down approaches for genotoxicity prediction.
First top-down approach
A workflow from the earlier publication “A transcriptomics-based in vitro assay for predicting chemical genotoxicity in vivo” by Magkoufopoulou et al. (2012) was developed, thereby reproducing their work as proof of principle. The workflow was created for one of the three approaches that were described in the study. There were some minor differences between the newly developed workflow and the original study, but overall the results of the original study were reproduced.
The workflow created using the Snakemake workflow manager is available from a GitLab software repository, where every step is clearly described in Snakefile to reproduce the approach described by Magkoufopoulou et al. (2012) and was used as reference for the transfer into an OpenRiskNet-based solution. The repository also includes required scripts as well as description of the steps necessary to reproduce the results.
The Snakemake-based workflow was converted to a Nextflow-based workflow, named nf-toxomix, in order to make use of the harmonization and interoperability of OpenRiskNet. The Nextflow version uses containerised steps, thus making it easier to deploy on any cloud infrastructure, and applicable to OpenRiskNet Virtual Environments. Furthermore, the Nextflow-based workflow has been translated into a more generic approach so that it can be applied to other toxicogenomics studies.
Second top-down approach
In this approach, transcriptomics data on human, mouse and rat in vitro liver cell models exposed to hundreds of compounds were collected from the diXa data warehouse, NCBI GEO and EBI’s ArrayExpress using the workflow from the first top-down approach. The obtained datasets were merged per species. This was done manually because of differences in the description of the datasets, e.g. differences in used ontologies, different metadata file formats. For all the compounds used in the experiments genotoxic and carcinogenic information was gathered from literature and several databases, including ToxPlanet. After normalization of the transcriptomics data (per species) and gathering of the genotoxicity/carcinogenicity information, the data were ready to be fed into the prediction models of ModelRX.
The transcriptomics data from the human, mouse, rat in vitro liver cell models and the toxicological information are available through OpenRiskNet (link).
Currently available services:
-
Our platform searches the content from 500+ websites and quickly delivers relevant chemical hazard and toxicology data.Service type: Database / data source, Data mining tool, Service
-
Service type: Database / data source
-
Processed data (counts and fold changes) based on the transcriptomics data provided by Open TG-GATEs and DrugMatrixService type: Database / data source
-
A database for curated toxicogenomic datasetsService type: Database / data source, Application, Visualisation tool, Software
-
Service to run Nextflow pipelinesService type: Software, Service, Workflow tool
-
Interactive computing and workflows sharingService type: Visualisation tool, Helper tool, Software, Analysis tool, Processing tool, Workflow tool
-
Computation research made simple and reproducibleService type: Database / data source, Visualisation tool, Software, Analysis tool, Service, Workflow tool
Related resources
In the past few decades, major initiatives have been launched around the world to address chemical safety testing. These efforts aim to innovate and improve the efficacy of existing methods with the long-term goal of developing new risk assessment paradigms. The transcriptomic and toxicological profiling of mammalian cells has resulted in the creation of multiple toxicogenomic datasets and corresponding tools for analysis. To enable easy access and analysis of these valuable toxicogenomic data, we have developed ToxicoDB (toxicodb.ca), a free and open cloud-based platform integrating data from large in vitro toxicogenomic studies, including gene expression profiles of primary human and rat hepatocytes treated with 231 potential toxicants. To efficiently mine these complex toxicogenomic data, ToxicoDB provides users with harmonized chemical annotations, time- and dose-dependent plots of compounds across datasets, as well as the toxicity-related pathway analysis. The data in ToxicoDB have been generated using our open-source R package, ToxicoGx (github.com/bhklab/ToxicoGx). Altogether, ToxicoDB provides a streamlined process for mining highly organized, curated, and accessible toxicogenomic data that can be ultimately applied to preclinical toxicity studies and further our understanding of adverse outcomes.
The OpenRiskNet case studies (originally outlined in Deliverable 1.3) were developed to demonstrate the modularised application of interoperable and interlinked workflows. These workflows were designed to address specific aspects required to inform on the potential of a compound to be toxic to humans and to eventually perform a risk assessment analysis. While each case study targets a specific area including data collection, kinetics modelling, omics data and Quantitative Structure Activity Relationships (QSAR), together they address a more complete risk assessment framework. Additionally, the modules here are fine-tuned for the utilisation and application of new approach methodologies (NAMs) in order to accelerate the replacement of animals in risk assessment scenarios. These case studies guided the selection of data sources and tools for integration and acted as examples to demonstrate the OpenRiskNet achievements to improve the level of the corresponding APIs with respect to harmonisation of the API endpoints, service description and semantic annotation.
In this case study a transcriptomics-based hazard prediction model for identification of specific molecular initiating events (MIE) was foreseen based on (A) top-down and (B) bottom-up approaches. The MIEs can include, but are not limited to: (1) Genotoxicity (p53 activation), (2) Oxidative stress (Nrf2 activation), (3) Endoplasmic Reticulum Stress (unfolded protein response), (4) Dioxin-like activity (AhR receptor activation), (5) HIF1 alpha activation and (6) Nuclear receptor activation (e.g. for endocrine disruption). This case study focussed on two top-down approaches for genotoxicity prediction. The first approach resulted in the creation of a Nextflow-based workflow from the publication “A transcriptomics-based in vitro assay for predicting chemical genotoxicity in vivo” by Magkoufopoulou et al. (2012), thereby reproducing their work as proof of principle. The Nextflow-based workflow has been translated into a more generic approach, especially for step 1, forming the basis of the second top-down approach. In this approach transcriptomics data together with toxicological compound information were collected from multiple toxicogenomics studies and used for building a metadata genotoxicity prediction model.
Report
Transcriptomics data from human, mouse, rat in vitro liver models
The OpenRiskNet project (https://openrisknet.org/) is funded by the H2020-EINFRA-22-2016 Programme. Here we present how the concept of Adverse Outcome Pathways (AOPs), which captures mechanistic knowledge from a chemical exposure causing a Molecular Initiating Event (MIE), through Key Events (KEs) towards an Adverse Outcome (AO), can be extended with additional knowledge by using tools and data available through the OpenRiskNet e-Infrastructure. This poster describes how the case study of AOPLink, together with DataCure, TGX, and SysGroup, can utilize the AOP framework for knowledge and data integration to support risk assessments. AOPLink involves the integration of knowledge captured in AOPs with additional data sources and experimental data from DataCure. TGX feeds this integration with prediction models of the MIE of such AOPs using either gene expression data or knowledge about stress response pathways. This is complemented by SysGroup, which is about the grouping of chemical compounds based on structural similarity and mode of action based on omics data. Therefore, the combination of these case studies extends the AOP knowledge and allows biological pathway analysis in the context of AOPs, by combining experimental data and the molecular knowledge that is captured in KEs of AOPs.
ToxTargetLinks
This report details the work involved in the federation of compute and data resources between the OpenRiskNet e-infrastructure and external resources. The reference environment has been designed to be capable of handling the majority of requirements for users’ wishes to deploy and run services. However specific situations demand solutions where either the computation, the data or both reside outside the OpenRiskNet e-infrastructure. This deliverable is related to Tasks 2.7 (Interconnecting virtual environment with external infrastructures) and Tasks 2.8 (Federation between virtual environments). Resource intensive analyses, such as those performed in toxicogenomics, can have CPU, memory or disk requirements that cannot be assumed to be available across all deployment scenarios. Human sequencing data may have restrictions on where it can be processed and the vast quantity of this data often predicates that it is more efficient to “bring the computation to the data”. In achieving Tasks 2.7 and 2.8, we can demonstrate how the virtual environment can utilise external infrastructure including commercial cloud providers and data stores.
Slides
Whole genome transcriptional profiling allows global measurement of gene expression changes induced by particular experimental conditions. Toxic treatments of biological systems, such as cell models, may perturb interactions among genes and, in toxicogenomics, such perturbations assessed by transcriptional profiling are used to predict impact of toxic compounds. Form early days on, this toxicogenomics-based approach for predicting apical toxicities, has been dedicated to the purpose of improving predictions of genotoxicity and carcinogenicity in vivo. Over the past decade large amounts of transcriptional profiling data have been generated for in vitro study models using various chemical compounds, across different doses and time points as well as different organisms. As part of the H2020 EU project OpenRiskNet, we propose a large-scale integrative analysis approach using these data sets for predicting genotoxicity and carcinogenicity in vivo. From the diXa Data Warehouse, NCBI GEO, and EBI ArrayExpress we collected gene expression data for human in vitro liver cell models exposed to 125 compounds with known genotoxicity information at different time points and dosages resulting in 822 experiments. We analyzed these data sets using ten different classification algorithms, thereby using 80% of the data for training and 20% for testing. Support Vector Machines algorithm had the best accuracy for predicting genotoxicity in vivo at 92.5% with 95% specificity and 87% sensitivity. Upon identifying deregulated gene-gene interaction networks by applying ConsensusPathDB, the top 5 of affected pathways are related to p53-centered pathways. The results from our meta-analysis demonstrate both high accuracy and robustness of transcriptomic profiling of genotoxicity hazards across a large set of genotoxicants and across multiple human liver cell models. We propose that these assays can be used for regulatory purposes, certainly when applied in combination with the traditional genotoxicity in vitro test battery. Next, we want to perform similar analyses on rat and mouse data and identify core orthologous genes among the three different species that are potential predictive targets for assessing genotoxicity and carcinogenicity across different biological systems.
JBayjanov_MetaAnalysis_EuroTox2018.pdf
Slides
Transcriptomics data from human, mouse, rat in vitro liver models